Artikelaktionen
EuroVR 2016
3D reconstruction with a markerless tracking method of flexible and modular molecular physical models: towards tangible interfaces
urn:nbn:de:0009-6-46956
Abstract
abstract Physical models have always been used in the field of molecular science as an understandable representation of complex molecules, particularly in chemistry. Even if physical models were recently completed by numerical in silico molecular visualizations which offer a wide range of molecular representations and rendering features, they are still involved in research work and teaching, because they are more suitable than virtual objects for manipulating and building molecular structures. In this paper, we present a markerless tracking method to construct a molecular virtual representation from a flexible and modular physical model. Our approach is based on a single RGB camera to reconstruct the physical model in interactive time in order to use it as a tangible interface, and thus benefits from both physical and virtual representations. This method was designed to require only a light virtual and augmented reality hardware setup, such as a smartphone or HMD & mounted camera, providing a markerless molecular tangible interface suitable for a classroom context or a classical biochemistry researcher desktop. The approach proposes a fast image processing algorithm based on color blob detection to extract 2D atom positions of a user-defined conformation in each frame of a video. A tracking algorithm recovers a set of 2D projected atom positions as an input of the 3D reconstruction stage, based on a Structure From Motion method. We tuned this method to robustly process a few key feature points and combine them within a global point cloud. Biological knowledge drives the final reconstruction, filling missing atoms to obtain the desired molecular conformation.
Keywords: Molecular Tangible Interface , , Physical model (Peppytide) , Structure from Motion , Markerless Tracking.
Keywords: Markerless Tracking, Molecular Tangible Interface, Physical model (Peppytide), Structure from Motion
:
Physical models have long been used to represent molecules and give a better perception of complex molecular structures like DNA and proteins. The first physical model by Pauling and Corey [ CP53 ] was static but scaled. Early physical models also contributed to study important molecular structures [ Hod49 ] (Figure 1).
It was only recently that flexibility, an important characteristic of the protein dynamic, has been introduced in physical molecular models. Peppytide [ CZ13 ] is a flexible, foldable, 3D-printed molecular model including magnets, able to accurately reproduce important features of the polypeptide chains such as bond lengths and angles between atoms. The rotational barriers within the protein backbone and a long-range energy is approximated thanks to magnets. This allows to create the hydrogen bonds inside the backbone which are needed for proteins to fold in secondary structures, mainly α-helix and β-sheet (Figure 2).
Figure 1. Hodgkin physical models of penicillin with patterns casted by X-rays (a) in 1953 and (b) in 1959.

Figure 2. Peppytide molecular flexible and modular model folded as (a) α-helix and as (b) β-sheet. The Peppytide model is made of amide pieces (in black) linked by an alpha carbon part (in white). The red and blue parts are one of the 20 different side chains of proteins. We built and painted a custom colored model (c) for RGB tracking purpose.

Moreover, the modularity of this model makes it an interesting tool for teaching biochemistry and protein folding concepts, two important basis of molecular biology courses.
While visualization software tools provide a wide range of dynamic and customizable 3D representations aiding the understanding of the bio-macromolecular complexity [ HDS96 , Sch18 , LTS13 ], physical models are still actively used as a research and teaching tool in the biochemistry field. Indeed the interaction with computer molecular models are usually performed with a 2D input device and require some form of manipulation metaphor. Noticeable work has been carried out in haptic manipulation of molecules in a virtual world [ Sto01 , FNM09 ] but it usually requires an expensive setup. Also, some manipulation issues explain why this approach is not widely adopted for teaching or research purposes: the interaction volume is restrained.
Physical and virtual molecular models are complementary in terms of interaction, manipulation and rendering, but only a few solutions combine the two representations. To assist the teaching of biochemistry and help researchers to study molecular processes, a molecular physical model steering a virtual representation would be an interesting asset. Thanks to the resulting tangible interface, the researcher would gain a direct interaction with the molecule and benefit from various pieces of information attached to the virtual representation.
Olson et al. [ GSSO05 ] address this issue with a static tracker-based tangible interface, augmented with relevant molecular information. Although trackers present different benefits (accurate tracking, inexpensive setup), they also present some significant drawbacks. The trackers occupy a significant physical space that breaks the physical molecular representation. Moreover, markers are not suited for flexible and modular models, especially during building and manipulation, because of possible hand occlusions (Figure 3).
Figure 3. 3D-printed molecular model with markers to add information via augmentation (from [ GSSO05 ]).
Figure 4. Modular tangible input device for character animation (from [ JPG14 ]). With its embedded sensors, this physical prop is used to precisely drive a virtual object.

Recently, a notable progress has been made in character animation [ JPG14 ] where pluggable parts with embedded sensors are linked to each other to drive the behaviour of a virtual animated character (Figure 4). Thanks to these sensors, the movements of the physical parts are accurately reproduced in the virtual world and occlusion issues are alleviated. However, this requires a significant electronic system with a power supply and connectors between parts. Even if a battery could power each module, a major miniaturization effort would be necessary to fit all the necessary electronic parts inside a small modular physical model.
A markerless tracking method combined with a full numerical reconstruction would avoid these drawbacks and gain the realistic flexibility of a modular and foldable physical model.
We thus introduce an original and lightweight markerless tracking method to reconstruct a 3D, flexible, modular molecular physical model using a single RGB camera (Figure 5).
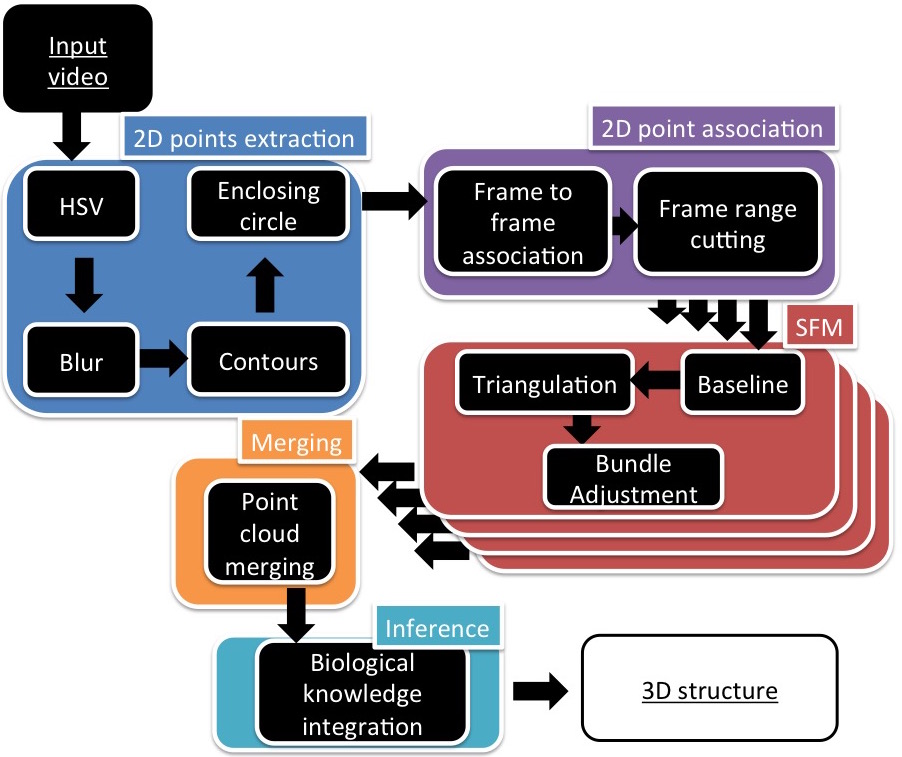
From a colored physical model in a given conformation, the first stage of this method is a fast image processing algorithm that extracts 2D positions of atoms, coupled with a tracking algorithm to associate them throughout the frames of a video input stream. Because tracking all the atoms in all the frames is unrealistic, the result of this stage is a series of incomplete tracking in video sub-sequences. A Structure From Motion (SFM) method was designed and tuned to process the sparse but robust 2D feature points tracked in the previous stage, providing a set of partial point clouds. A merging algorithm based on atom colors is then applied to combine all the partial points clouds into a single, coherent one. Eventually biological knowledge is exploited to complete the 3D reconstruction and to obtain the final peptide conformation.
The challenge is to track a modular model with a single RGB camera without using trackers. Thus the input consists in a color video stream processed in interactive time.
The stream is expected to contain a set of continuous viewpoints of a colored Peppytide model in a fixed conformation. The Peppytide model does not specifically define atom colors so we simplified the image processing step by assigning a color scheme, based on the commonly used Pymol color scheme [ Sch18 ]. The only modification made was to paint the hydrogen atoms using yellow to be readily tracked.
Firstly, in order to separate colors and be less sensitive to brightness variations, each frame of the video is processed in the HSV (Hue-Saturation-Value) color space. A mask for each color is created and can be processed independently from the others. As only a small number of atom types are represented in proteins, there is no color ambiguity and they all can be processed in parallel.
As a pre-processing step, noise removal with classical erosion and dilation morphological operations is applied. A Gaussian blur is used to enhance the Suzuki [ SA85 ] border following detection algorithm.
Finally, an iterative minimal enclosing circle algorithm is applied on each border, computing a 2D circle position, a projected radius and the nature (color) of each visible atom. These circles are then filtered by a radius and color thresholds to remove remaining artifacts.
These thresholds can be passed as parameters of the program or can be set by asking the user to interactively select an atom of each color in a frame of the video to tune them with the user model. The method was implemented in C++ using the OpenCV 2.4.10 library for the image processing part. Using the GPGPU implementations of some OpenCV algorithms and rewritting the HSV range selection algorithm in CUDA allowed us to run the image processing step in interactive times.
Figure 6. Sample of an execution showing the number of detected atom blobs by frame. As the model is rotating some atoms are lost, while others appear.
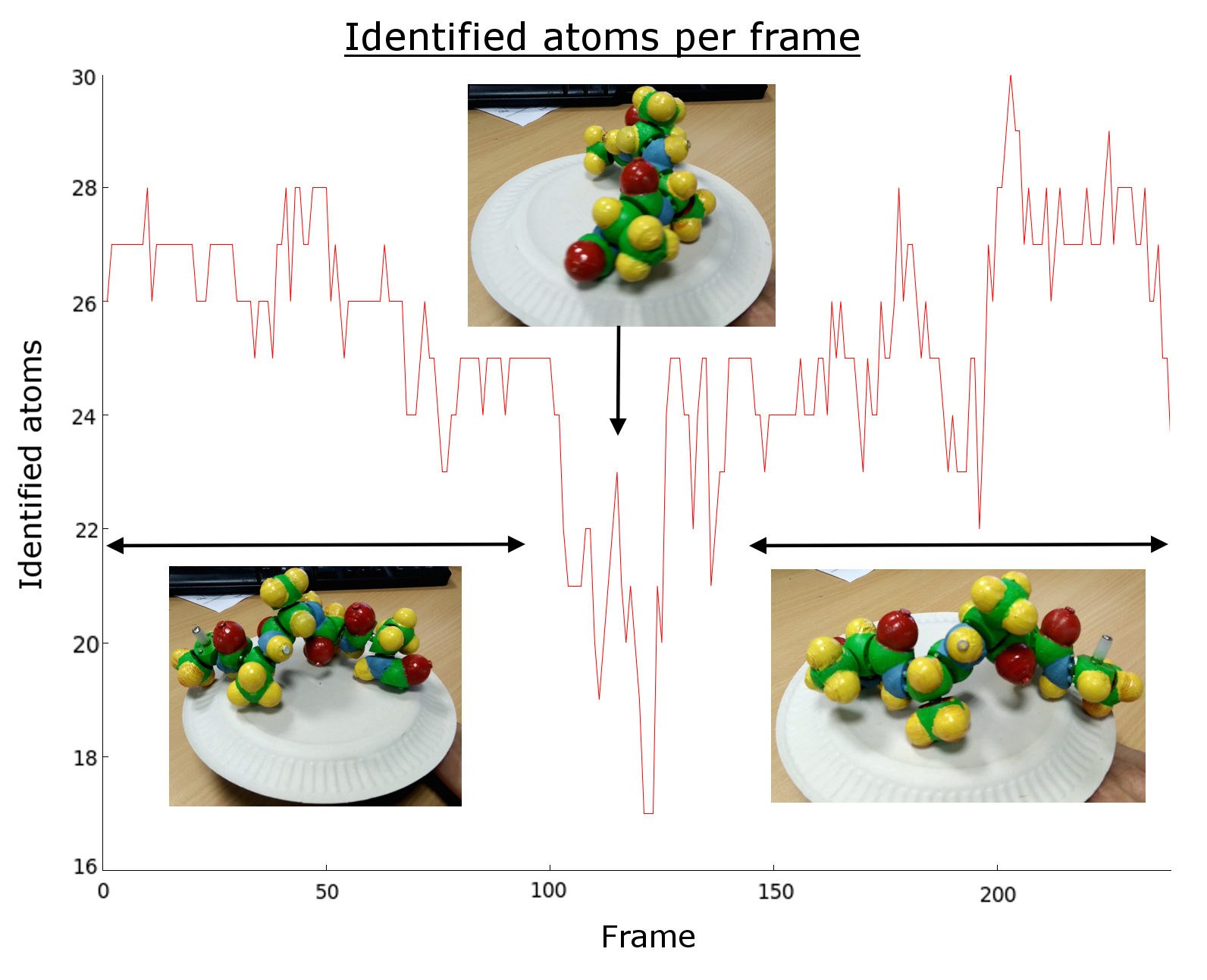
One of the key step of this approach is the association of the 2D atom center feature points coming from the processing of each image of the video. The goal is to minimize the number of incorrect associations which are highly penalizing the subsequent Structure From Motion step. The number of feature points detected in each frame drops when the physical model is viewed under an unfavorable angle (Figure 6). This expected behavior will severely impact the tracking algorithm as a lot of information is lost during these critical frames. Unlike usual reconstruction methods based on standard tracking algorithms, there is no way to keep track of some points to get a continuous association between points of the previous frames and new ones.
Consequently, the input video is cut into shorter sub-sequences, the cuts intervening when the number of atoms tracked in the range falls below a threshold. This value is directly linked to the minimum number of points needed to perform the SFM step.
Starting from the first frame, each atom is compared with same color atoms of the next frame using the radius variation and the position variation. From the second to the last frame, no new atom is created to increase the robustness of the tracking, which means that the number of points tracked in the video will decrease from beginning to end. This is done to identify ranges of frames in which the number of tracked points is nearly constant therefore to obtain a reconstruction of a part of the protein. The idea is to complete and merge parts using biochemical knowledge in a forthcoming step.
Note that the final number of atoms depends on the number of identified atoms of the first frame. Thus it is best to choose the frame with the maximum number of identified atoms in the video. A backtracking step is performed to find atoms that have been missed in a frame or more: for each frame, atoms are compared with the last N frames with slightly increased radius and motion thresholds.
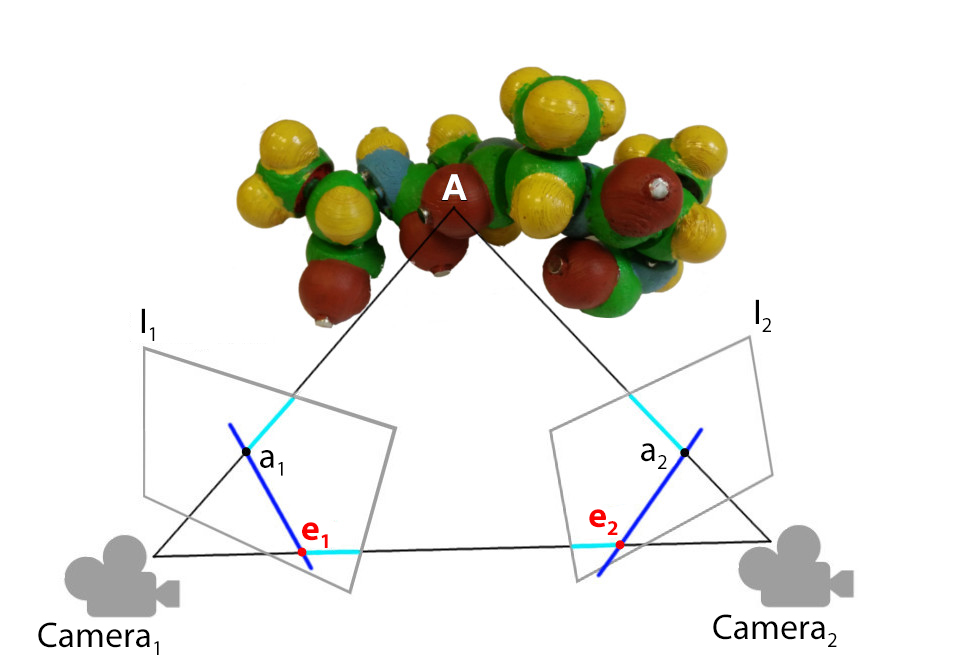
Figure 7. As explained in [ THWS10 ], using the pinhole camera assumption, the point A is projected on the image plane of the camera Camera 1 and Camera 2. The information that a 1 in the frame 1 corresponds to a 2 in the frame 2 allows to project these points during the SFM. Although the projection lines should intersect at A in theory, in practice the lines do not cross. Using the epipolar geometry, the points have to be triangulated to get an approximation of A.
For each range of frames determined during the tracking step, a number of atoms (consisting in sets of 2D points with associated color codes) are tracked from the beginning to the end of the range. The Structure From Motion (SFM) method takes these 2D points as inputs and outputs a 3D point cloud (Figure 7).
Figure 8. OpenCV ORB feature points matching between two frames of a video of the moving physical model. Specularities, shadows and links between atoms are the only points tracked. Moreover, points are not uniformly distributed over the protein.

The SFM method relies on the epipolar geometry to compute the depth of object points. Epipolar geometry defines the relation between two frames, taken from different point of views, that links the position and orientation of the camera, usually using a Pinhole camera model (perspective projection).
In Figure 7, the point A is projected on a 1 on the image I 1 of Camera 1 and on a 2 on the image I 2 of Camera 2. Epipoles e 1 and e 2 are the projections of the camera center on the image plane of the other camera. Epipolar lines are the lines that link the projections of 3D object points to the epipole ( e 1 to a 1 and e 2 to a 2, in dark blue), they give the association between projected points on different cameras. With this information, the fundamental matrix can be computed to obtain an algebraic form of the epipolar constraints. Thanks to the fundamental matrix, the position and the pose of each camera can be computed besides the depth of the object 2D points.
Even if the SFM method can reconstruct as few as 4 points in a 3 viewpoints set [ Ull79 ], the usual setup makes use of numerous feature points with algorithms like SURF [ BTG06 ] or ORB [ RRKB11 ].
Feature points extraction is not well suited in our case as it does not focus on valuable information of the physical models but instead on details like specularities and shadows (Figure 8). Moreover, the main information to be extracted from each frame is the position of each atom center and not the general shape of the protein.
In our method, only a few feature points can be reliably extracted from the video, so the SFM method is modified accordingly. The baseline triangulation is chosen between two frames separated by a fixed number of frames, chosen to be a 5 frames interval in the same sub-range.
If the fundamental matrix computation (obtained with the 8 points algorithm [ Har97 ] proposed in OpenCV ) returns an acceptable result, 2D points are triangulated. The mean re-projection error - that is the mean difference in position between the projected points and the 2D input points - is used as the decisive value.
The quality of the baseline triangulation will significantly affect the final reconstruction as every frame will modify and possibly add some points to the baseline point cloud. This iterative step is using every frame of the range, estimating a pose of the camera, triangulating and adding some points if not present in the point cloud.
The final standard bundle adjustment (BA) step is performed every time a frame is used to refine the point cloud. It consists of a minimization of the re-projection error by slightly changing the 3D points and optionally the camera matrices. The SSBA library was used to compute the bundle adjustment step [ Zac14 ].
The point clouds emanating from the SFM frame ranges do not always share common atoms that could be used to compute a unique transformation and a unified set of points. When two point clouds describe different parts of the model, they have to be assembled into one and the classical approach is to use the Iterative Closest Point algorithm (ICP) [ BM92 ]. Like SFM, a large set of points is expected in the usual application of the ICP method and the two point clouds generally overlap significantly. In our approach, the point clouds are sparse and poorly overlap, but color information can help overcome this problem.
The proposed algorithm takes the two point clouds (each point with a given color) and a set of overlapping points, if they have been identified. The two point clouds are expected to have the same scale. If no corresponding points have been identified, the procedure searches for some with the following algorithm: for each couple of points of the first point cloud (pc 1) search a similar duet with the same colors in the second point cloud (pc 2) according to a distance criterion. For each found duet of pc 1, add a new point not in the duet to form a triplet and look for a similar triplet in pc 2. Finally, evaluate the best rigid transformation with a classical SVD method and score all the solutions based on the number of points of pc 1 with the same color and approximate position as pc 2 points.
If one or two overlapping points are available, the approach consists in minimizing a function that is minimum when all the pc 1 points overlap pc 2 points, as a function of the rigid transformation between the two sets. A simple sampling of the initial pose is used to avoid local minima.
Obviously, the more information available to the algorithm, the more accurate the merged point cloud will be.
Once the point clouds have been merged into one, some atoms of the protein are still missing because of several types of occlusions (e.g. self occlusions or hand occlusions) during the manipulation. Tracking errors are also possible, resulting in points being rejected during the SFM stage. To solve this issue and because biochemistry constrains the system, biochemical knowledge can help remove some uncertainties about missing atom positions.
The first step is to identify complete or partial model parts in the point cloud based on angles between points of the part. Amide parts, one of the two main parts of the physical model, are composed of 4 atoms. We propose to use angles between three atoms of the same part and compare them to known possible values. The intrinsic scale factor of the model is also deduced from this information as a mean of the scale factor of the different amide parts identified.
Proteins are composed of amino-acids that are represented as a group of 4 parts in the Peppytide model: 2 amide parts linked by the alpha-carbon piece in the middle, holding the lateral chain part (Figure 9). As the protein is a chain of amino-acids, there is an order of each group of parts composing the chain. The second step consists in deducing the order of the identified pieces in the point cloud. It is done by comparing the distance between parts and deducing if the parts can be consecutive or are too far away to be linked. Maximum and minimum distances between amide parts were computed beforehand using the numerical Peppytide model. If enough parts are identified and with the sequence computed, it is then possible to deduce the total number of amino acids of the peptide ( i.e. the number of Peppytide parts) and therefore we can deduce the number of missing parts in the point cloud.
In a third step, because the Peppytide mechanical properties are available, a virtual reconstruction of the molecule is created based on the number of amino acids previously obtained with the deduced scale factor. Using a physical engine, attractive forces are applied between the associated parts in the point cloud and the virtual model. The mechanical constraints stored inside the virtual representation of Peppytide (including collisions and hinge constraints between rigid bodies) realistically drive the virtual model inside the point cloud and give a final reconstructed structure of the peptide. As parts are attracted to an ideal position, this step can be seen as a minimization with physical constraints given by the Peppytide model. This step was implemented in the Unity3D game engine. The numerical model was created with its mechanical properties in Unity, benefiting from the built-in PhysX physical engine.
Some form of ground-truth measurement of the observed model is necessary for the evaluation of the reconstructed molecule. Even with a 3D-scanner, extracting 3D atom positions from the real world is a difficult task. We propose a way to obtain an approximation of the reference structure using the φ and ψ angles given by magnet positions. In proteins, these dihedral angles define the backbone conformation and, in the Peppytide model, magnets are placed to reproduce several average protein conformations of known protein structures.
Figure 9. (a) Numerical representation of Peppytide with Psi angle in black and Phi angle in gray. (b) Magnet configurations in the Peppytide model give specific Phi and Psi angles.
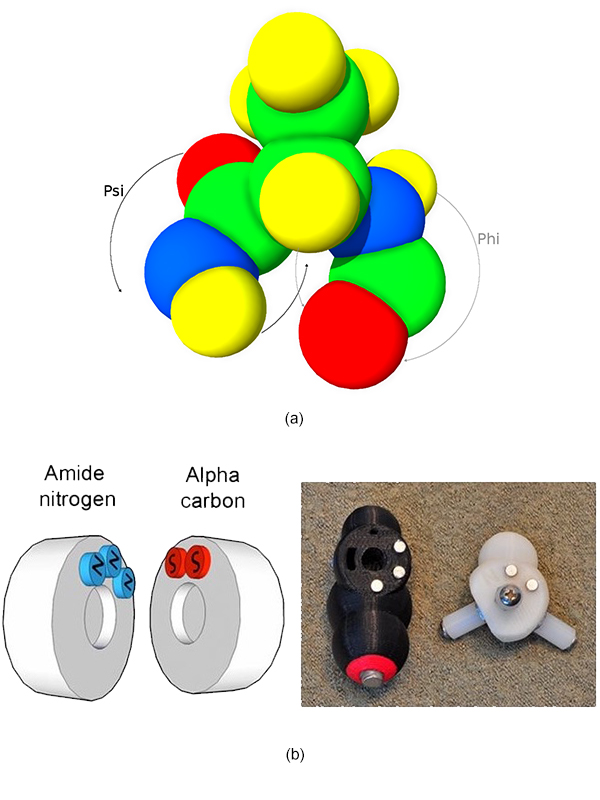
The Peppytide model defines two magnet configurations for the ψ angle and four for the φ angle. Knowing magnet conformations implies knowing every φ and ψ angles of the peptide. As the model is made of articulated rigid parts, we can compute every atom position except for the lateral chains.
A virtual representation of the reference structure can then be created and compared with the proposed solution. Once the reconstructed structure is converted in Ångström using the Peppytide scale factor, we compute a classical measure in structural biology called RMSD (Root-Mean-Square Deviation) to give an idea of the differences between the proposed solution and the reference structure.
For each amino acid in the Peppytide model there are 8 (φ/ψ) angle couples. For 5 amino acids, there are therefore 85 = 32768 (φ/ψ) angle couples. As this evaluation method contains manual steps (assembling the peptide, identifying the φ and ψ angles for each amino acid and filming the video), it is not possible to test such a large set of conformations. We focused on testing three common secondary structure conformations: one β-sheet, an unfolded structure with 5 amino acids, one highly compact α-helix of 6 amino acids and one moderately compact conformation (5 amino acids).
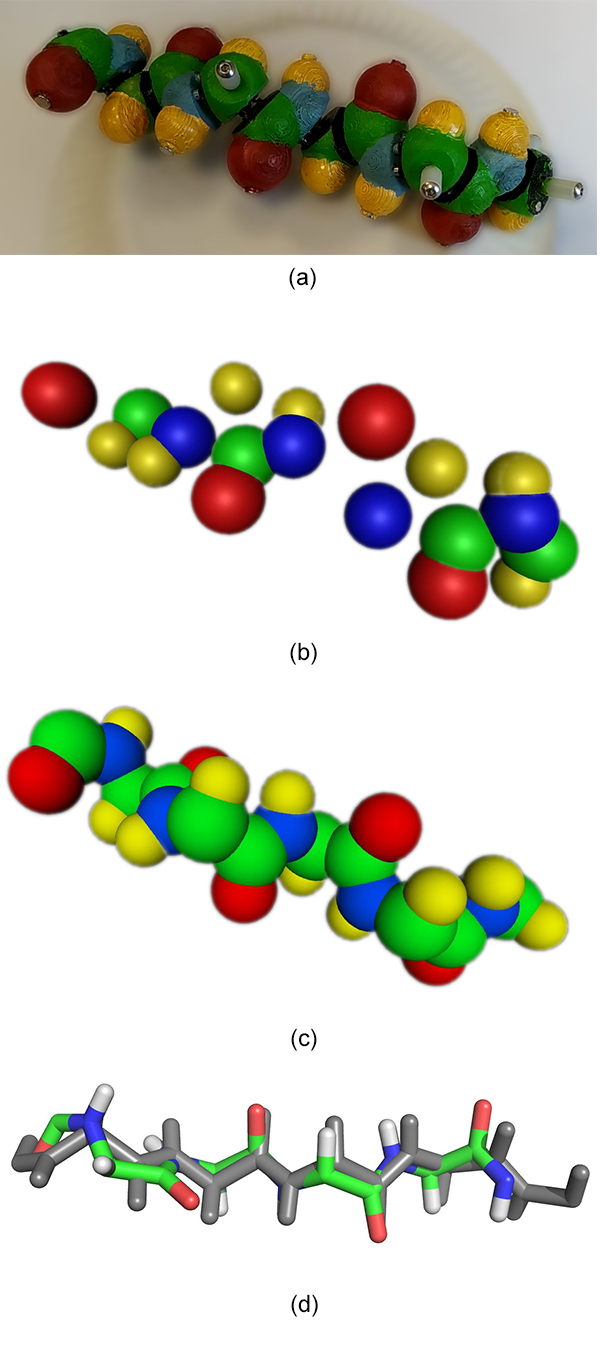
The best-case scenario for our technique is an unfolded structure where every atom is clearly visible and easy to track. One strand of a β-sheet structure, a common conformation in proteins, corresponds to this unfolded state. As expected, applying our method to a video of a 5-amino-acid peptide in this conformation leads to a full reconstruction in the end. 11 atoms of the initial 30 atoms are missing in the unique point cloud output (Figure 10b) but it is sufficient to identify the number of amino acids and fill the missing atoms thanks to biochemical knowledge (Figure 10c). The alignment with a reference structure gives a 0.889Å RMSD in this example (Figure 10d). The scale factor of Peppytide parts is 1Å = 0.268" = 0.35mm, the error relative to the physical model dimensions is about 8.3mm.
Figure 10. (a) Input video frame of the 5-amino-acid β-sheet peptide without lateral chains. (b) One output point cloud after the SFM step. (c) Full biological knowledge-driven reconstruction, the correct number of amino acids leads to the same atom count. (d) Reference structure in gray is aligned with the reconstructed structure with a RMSD of 0.889Å.
We created a more compact structure to test the method on an occluded structure in which some atoms are hidden (Figure 11a).
Figure 11. (a) Input video frame of the 5 amino acid peptide with lateral chains. (b) One output point cloud after the SFM step. (c) Full biological knowledge driven reconstruction.
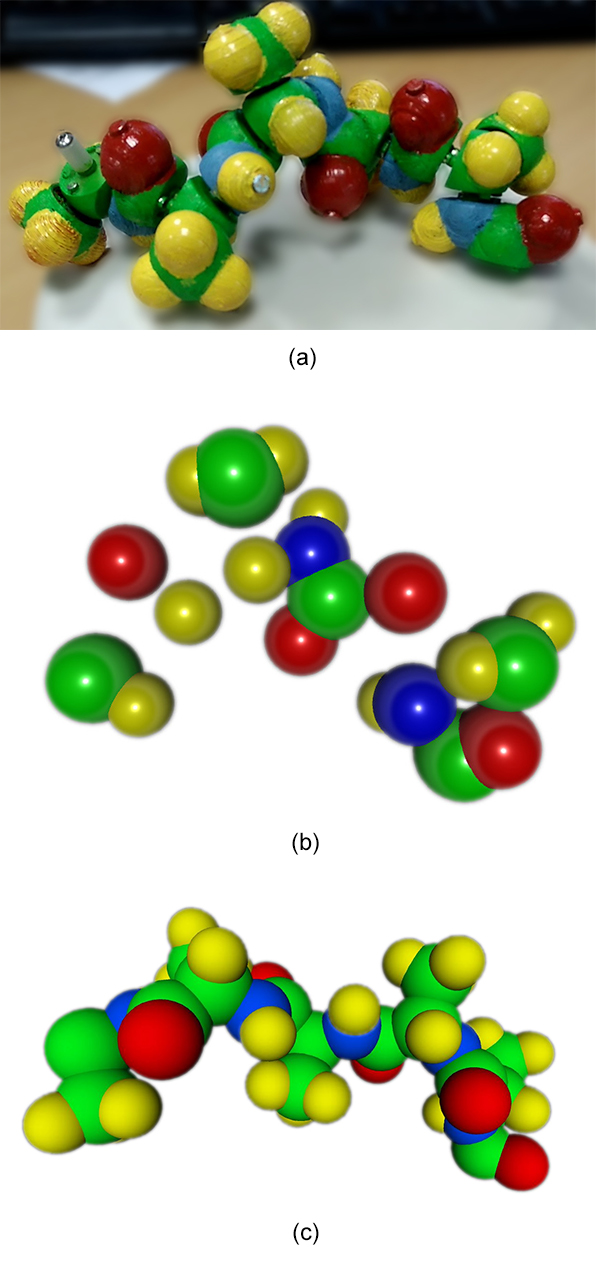
On the initial 50 atoms, only 16 valid points are triangulated in this point cloud, then processed to give a full reconstruction. The RMSD value of the atom backbones in the example presented in Figure 11c is 1.07Å that is 10mm in world space dimensions. All atoms have been reconstructed but only two amide parts are misplaced, inducing wrong φ and/or ψ angles. As expected, parts with less information in the point cloud tend to induce atom misplacement in the reconstruction even if the final 3D reconstruction remains chemically correct thanks to Peppytide properties.
The RMSD is prone to accentuate small dissimilarities. That is the reason why the world space translated error of 10mm is high. The error is distributed over all the alignment.
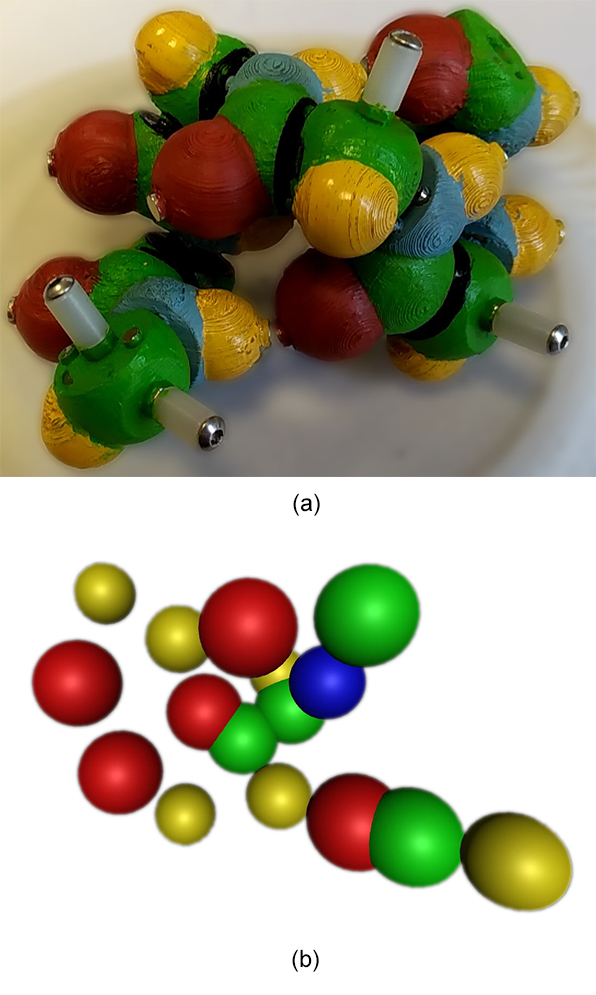
Finally, our method was applied on a highly compact structure that is biologically relevant, an α-helix of 6 amino acids (Figure 12a). This structure is challenging for our approach because most of the atoms are occluding others, so that the image processing step will not capture all the important parts of the peptide. A point cloud is indeed generated (Figure 12b) but the noise inducing position fluctuations and the absence of important atoms do not allow to obtain the correct number of amino acids nor the sequence of parts in the peptide chain.
Figure 12. (a) Input video frame of a 6-amino-acid α-helix peptide without lateral chains. (b) One output point cloud after the SFM step.
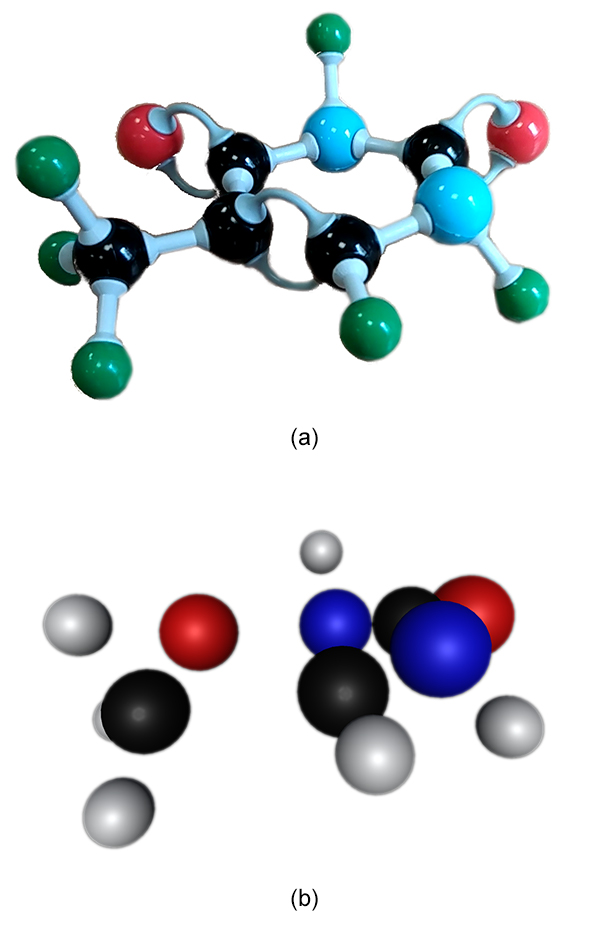
MolyMod is a physical model used to teach chemistry. Applying our method to this model is straightforward. Green spheres were used to represent hydrogens to optimize the tracking step. As the atoms are distant from each other, the color-based tracking correctly gives 2D positions in nearly all frames (Figure 13) and the SFM step usually outputs a single point cloud. The Figure 14 shows an example of the application of the method on a thymine DNA base containing 15 atoms, 13 of which are reconstructed in this example. In a similar way as it was done with Peppytide, chemical knowledge could be used to recover the 2 missing atoms, to further precise the atom positions and obtain a full 3D point cloud.
Figure 13. The number of tracked atoms in each frame of a video of a thymine (15 atoms) using the MolyMod physical model shows that it is robustly tracked using our image processing step, all atoms can be identified and few artifacts are present (more than 15 atoms).
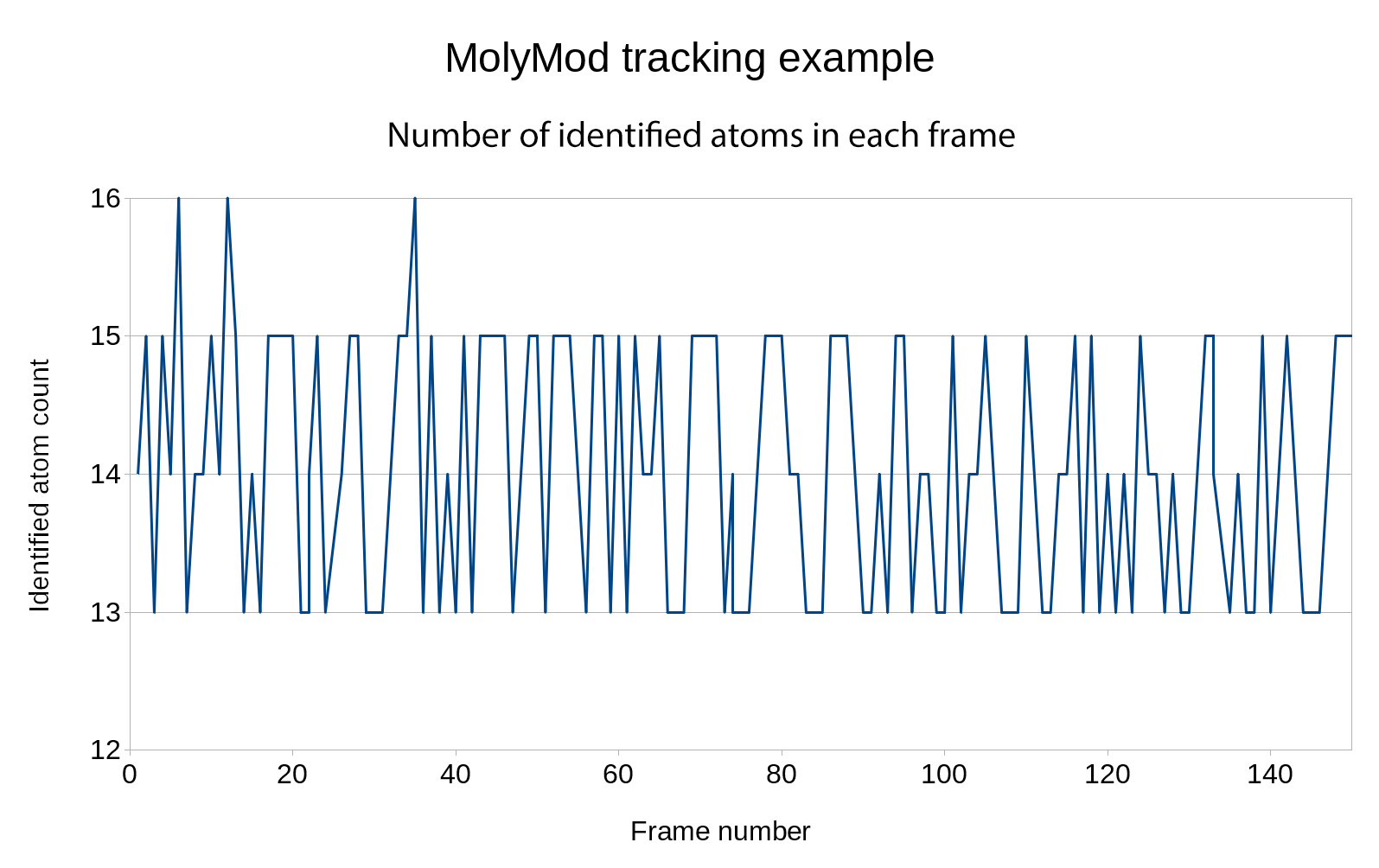
Figure 14. (a) Frame of the input video of the MolyMod physical model assembled as a thymine, the DNA base with green atoms as hydrogen. (b) Point cloud output of the method with hydrogen being colored in white. Only 2 carbon atoms are missing in the reconstruction and can be restored using chemical knowledge.
For now, our method can reconstruct a small protein from its static physical model. This is only the first step towards markerless tracking of molecular tangible interfaces, the next one being to reconstruct a moving flexible model manipulated in interactive time. In accordance with the weight and size of the physical model, the proposed approach is also limited to small peptide reconstruction.
Applying the method to another physical model named MolyMod, which is largely used to teach chemistry, showed that this approach is relevant for other models and can lead to direct applications addressing education challenges to effectively teach chemistry using augmented reality and physical models [ Che06 ]. An experiment is being conducted within a serious game setup: the student has to physically create a molecule and verify that it was built correctly by simply shooting a video of the molecular model with a smartphone. The tracking of chemistry-oriented physical models is less challenging because, most of the time, all atoms are separated and do not occlude one another.
Nevertheless, reconstructing a highly self-occluded and hand-occluded physical model can be troublesome. That is why a mixed approach with Hall-effect rotation sensors is being investigated to solve this issue. As the model is highly modular and because the interface should be easy to setup, we believe it should be wireless. By slightly increasing the scale of the Peppytide model, one could fit a small battery inside each part. If needed, a connector could replace the screw-based links to carry information and power. A small chip could provide the wireless connection to send rotation data to the computer. Image processing steps are still required in an augmented reality context to obtain the translations and augment the physical model even if an accelerometer is embedded. Although this would be pricier and harder to build, the accuracy of this tangible interface would open a wider range of application, especially in the research field.
On the other hand, preliminary tests have been conducted using a stereo camera or a RGB-D camera instead of a single RGB camera to get an instant accurate approximation of the depth of each pixel. As depth sensors tend to be integrated in smartphone cameras (as in Tango project by Google for instance), these devices should increase the reconstruction accuracy without implying a heavier setup. Apart from the depth sensor range not matching our small object sizes and infrared light being poorly reflected by the spheres, the registration algorithms to reconstruct a point cloud fail to identify the same points in different view points leading to an accumulation of noise at this point (Figure 15). This observation was valid for both Tango phone depth sensor (a Lenovo Phab 2 Pro device in our case) and a higher quality SR300 RealSense camera. Nevertheless, using a depth sensor can improve camera tracking and 3D reconstruction in several ways. Firstly, it can be used to distinguish color blobs merged together in 2D but with different depths, an important feature when reconstructing larger or packed molecules. Secondly, having an approximation of the depth for each identified atom in 2D could set the confidence interval for the estimated depth during the SFM step.
Thirdly, considering that raw depth maps can provide high-quality depth values (see Figure 15a), by using 2D atom matching between frames, we could obtain a precise estimation of the depth of each identified atom in every frame and compute a mean of these values by frame sampling. This option presents the benefit of being computationally light and does not require SFM nor ICP computations.
Figure 15. Different 3D scans using (a) & (b) a Tango phone and (c) an Intel RealSense camera. (a) A quick scan without moving around the molecule shows that the depth data are usable even for small objects. (b) The registration algorithm fails to associate points in different points of view. (c) The RealSense SDK provides a mesh of the scanned object but similarly, 3D points are poorly registered.

We proposed a markerless tracking approach using a single RGB video stream to reconstruct digital representations of tangible molecular interfaces based on the Peppytide model. The physical model was tracked using an image processing pipeline built with standard tools available in the OpenCV library. In addition, a fast SFM method was especially implemented and tuned to deal with reduced sets of points and designed for low computational resources. To achieve this process, biochemical knowledge was used to make further assumptions about the molecular structure and recover missing atom positions. This drives the final reconstruction stage to obtain a chemically relevant structure.
Furthermore, we are currently working on a mixed reality application of our approach in a biochemistry teaching context. Indeed, as the method does not require a restrictive setup and could use a single RGB webcam or a smartphone camera, the virtual reconstruction of this molecular model is widely accessible, targeting both researchers and teachers. We are thus currently performing an evaluation of the added value of our approach for teaching biochemistry, and we plan to assist researchers by providing tools based on tangible interfaces to interact with a numerical molecular simulation.
Besides, we propose several ways to improve 3D reconstruction of flexible and modular physical models with RGB-D cameras or embedded sensors, to go towards dynamic tangible interfaces and manipulate complex objects in VR & AR contexts.
This work was made possible, in part, by the support of the French government through a grant of the French National Research Agency (Equipex program) with reference ANR-10-EQPX-01. The authors wish to thank Promita Chakraborty for kindly providing the 3D-printing data to recreate the interface at the DIGISCOPE FabLab with Romain Di Vozzo's kind help.
[ BM92 ] Method for registration of 3-D shapes Proceedings SPIE 1611, Sensor Fusion IV: Control Paradigms and Data Structures, 1992 DOI 10.1117/12.57955, 0-8194-0748-8
[ BTVG06 ] Surf: Speeded up robust features Computer Vision - ECCV 2006, (A. Leonardis A. Pinz H. Bischof Eds.), 2006 Springer Berlin, Heidelberg pp. 404—417 DOI 10.1007/11744023_32, 978-3-540-33832-1
[ Che06 ] A study of comparing the use of augmented reality and physical models in chemistry education VRCIA '06 Proceedings of the 2006 ACM international conference on Virtual reality continuum and its applications, 2006 ACM New York, NY, USA pp. 369—372 DOI 10.1145/1128923.1128990, 1-59593-324-7
[ CP53 ] Molecular models of amino acids, peptides, and proteins Review of Scientific Instruments, 1953 8 621—627 DOI 10.1063/1.1770803, 0034-6748
[ CZ13 ] Coarse-grained, foldable, physical model of the polypeptide chain Proceedings of the National Academy of Sciences of the United States of America, 2013 33 13368—13373 DOI 10.1073/pnas.1305741110, 0027-8424
[ FNM09 ] Multisensory VR interaction for protein-docking in the CoRSAIRe project Virtual Reality 2009 4 273—293 DOI 10.1007/s10055-009-0136-z, 1359-4338
[ GSSO05 ] Tangible interfaces for structural molecular biology Structure 2005 3 483—491 DOI 10.1016/j.str.2005.01.009, 0969-2126
[ Har97 ] In defense of the eight-point algorithm IEEE Transactions on pattern analysis and machine intelligence, 1997 6 580—593 DOI 10.1109/34.601246, 0162-8828
[ HDS96 ] VMD: visual molecular dynamics Journal of molecular graphics 1996 1 33—38 DOI 10.1016/0263-7855(96)00018-5, 0263-7855
[ Hod49 ] The X-ray analysis of the structure of penicillin Advancement of Science 1949 22 85—89 0001-866X
[ JPG14 ] Tangible and modular input device for character articulation ACM Transactions on Graphics (TOG) 2014 4 Article no. 82, DOI 10.1145/2601097.2601112, 0730-0301
[ LTS13 ] Game on, science - how video game technology may help biologists tackle visualization challenges PLOS ONE 2013 3 Article no. e57990, DOI 10.1371/journal.pone.0057990, 1932-6203
[ RRKB11 ] ORB: An efficient alternative to SIFT or SURF 2011 International conference on computer vision (ICCV), 2011 pp. 2564—2571 DOI 10.1109/ICCV.2011.6126544, 978-1-4577-1101-5
[ SA85 ] Topological structural analysis of digitized binary images by border following Computer Vision, Graphics, and Image Processing 1985 1 32—46 DOI 10.1016/0734-189X(85)90016-7 0734-189X
[ Sto01 ] Haptic feedback: a brief history from telepresence to virtual reality Haptic Human-Computer Interaction, (Stephen Brewster Roderick Murray-Smith Eds.) 2001 Springer pp. 1—16 DOI 10.1007/3-540-44589-7_1, 978-3-540-42356-0
[ THWS10 ] Registration of sub-sequence and multi-camera reconstructions for camera motion estimation Journal of Virtual Reality and Broadcasting 2010 2 1—10 DOI 10.20385/1860-2037/7.2010.2, 1860-2037
[ Ull79 ] The interpretation of structure from motion Proceedings of the Royal Society of London B: Biological Sciences 1979 1153 405—787 DOI 10.1098/rspb.1979.0006, 0950-1193
[ Zac14 ] Robust bundle adjustment revisited Computer Vision - ECCV 2014, 2014 Springer Cham , pp. 772—787 DOI 10.1007/978-3-319-10602-1_50, 978-3-319-10601-4
Volltext ¶
-
 Volltext als PDF
(
Größe:
15.1 MB
)
Volltext als PDF
(
Größe:
15.1 MB
)
Lizenz ¶
Jedermann darf dieses Werk unter den Bedingungen der Digital Peer Publishing Lizenz elektronisch übermitteln und zum Download bereitstellen. Der Lizenztext ist im Internet unter der Adresse http://www.dipp.nrw.de/lizenzen/dppl/dppl/DPPL_v2_de_06-2004.html abrufbar.
Empfohlene Zitierweise ¶
Xavier Martinez, Nicolas Férey, Jean-Marc Vézien, and Patrick Bourdot, 3D reconstruction with a markerless tracking method of flexible and modular molecular physical models: towards tangible interfaces. Journal of Virtual Reality and Broadcasting, 14(2017), no. 2. (urn:nbn:de:0009-6-46956)
Bitte geben Sie beim Zitieren dieses Artikels die exakte URL und das Datum Ihres letzten Besuchs bei dieser Online-Adresse an.
-
 News
News